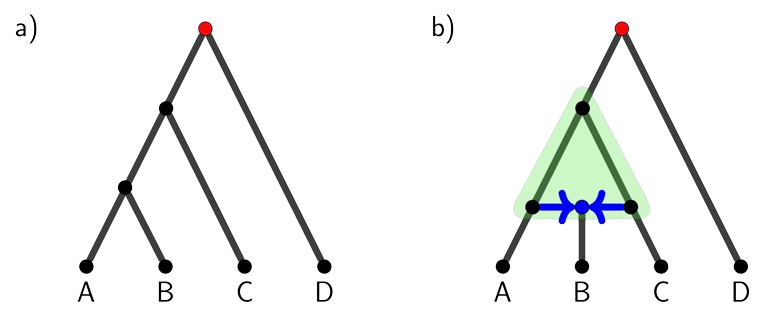
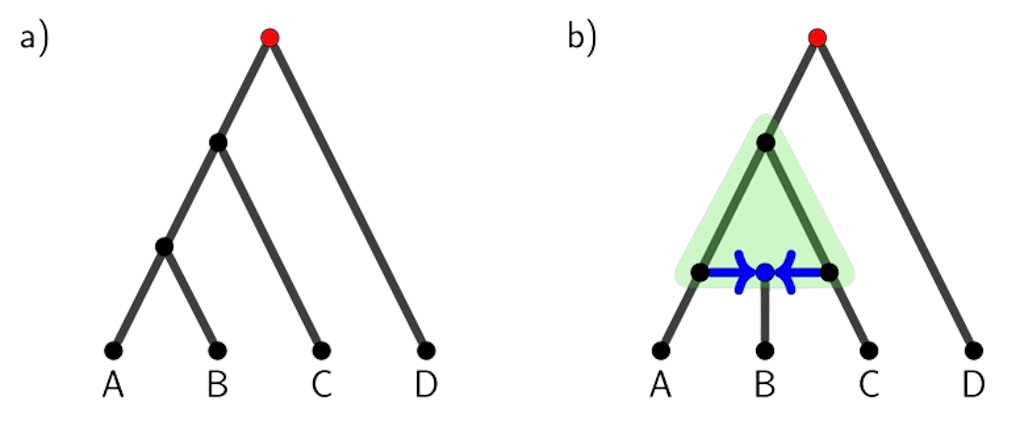
Phylogenomics is the study of how biological entities, such as species or populations, evolve through time using genomic data. These relationships are traditionally represented using bifurcating trees where each point of split represents a speciation event and the branch represents a lineage over time (Figure 1a). While the tree model depicts the historical relationships generally well, it sometimes oversimplifies the true complexity of evolution. This is because nature often does not follow a strictly branching pattern. For example, hybridization, a process of two distinct species interbreeding and combining their genetic material, can create evolutionary histories that are tangled and cannot be captured by a tree. These complex relationships are better represented by phylogenetic networks, which allow for both branching (i.e., speciation) and merging (i.e., hybridization) events in evolutionary history (Figure 1b). 17416_highlight_177.png140.57 KB
As high-throughput sequencing technologies advance, researchers now have access to large-scale genomic data across many species. This has opened up new opportunities to understand evolutionary relationships more precisely, but it has also introduced new computational challenges. Large datasets are powerful and full of biological signals, but they are also difficult to analyze efficiently. Accurately detecting hybridization events and reconstructing phylogenetic networks from these datasets requires new tools and algorithms that are both mathematically and statistically sound, as well as computationally scalable [1].
My long term research goal focuses on developing such tools. In particular, I work on creating methods that can reliably infer phylogenetic networks from genome-scale data, especially in cases where hybridization has played an important role in shaping contemporary species diversity. I am interested in building algorithms that are grounded in solid biological, statistical, and mathematical theory while also being practical enough to apply to real-world biological datasets. This means designing user-friendly methods that are not only accurate but also efficient in terms of computational time and memory usage.
A major obstacle is the lack of scalability. Many existing methods for inferring phylogenetic networks rely on a two-step process. First, they estimate gene trees from different parts of the genome. Then, they compare those gene trees to identify inconsistencies that suggest hybridization or other complex events. This approach has been useful, but it depends heavily on the accuracy of gene tree reconstruction. When sequences are short or the true evolutionary processes largely deviates from the existing models, gene trees can be very difficult to estimate correctly. This can introduce errors into the network inference and limit the reliability of the results.
As a first step to overcome these limitations, I developed a computational method called PhyNEST, which stands for Phylogenetic Network Estimation using Site Patterns [2]. It is an open-source software implemented in Julia, a modern programming language designed for high-performance scientific computing. PhyNEST works directly with DNA sequence alignments. Specifically, it analyzes small groups of four species at a time and records the patterns of nucleotides that appear at each site in the alignment. These patterns, known as quartet site patterns, carry important information about the underlying evolutionary relationships.
While the disadvantage of directly using sequence data is increased computational cost, PhyNEST employs a composite likelihood framework to speed up the analysis. Rather than attempting to compute the full likelihood of a complex network, which often becomes computationally infeasible for large datasets, PhyNEST calculates likelihoods for all quartets and then combines them to estimate the overall network. The composite likelihood framework offers a powerful balance between accuracy and efficiency [3]. Although it involves certain statistical approximations, it enables researchers to tackle far larger and more complex datasets than would be possible with full-likelihood methods.
A promising future direction for PhyNEST involves addressing key limitations to enhance its biological realism, computational performance, and empirical applicability. The current version of PhyNEST focuses on improving efficiency in network inference at the cost of accuracy by oversimplifying the underlying model. Future extensions of the model should be developed simultaneously with improvements in computational efficiency through algorithmic optimizations and parallel computation. In this way, the method can become more accurate without sacrificing efficiency. In addition, PhyNEST should expand its user-friendliness through a more intuitive interface and seamless integration with empirical data. These efforts will make PhyNEST more accessible to a broader community of researchers, including those with limited programming experience. Together, I believe these advancements will establish PhyNEST as a powerful and versatile tool for reconstructing complex evolutionary histories from genome-scale data.
[1] Kong S, Soíls-Lemus CR, and Tiley GP. (2025) Phylogenetic networks empower speciation biology. Proc. Natl. Acad. Sci. U.S.A 122:31 e2410934122 doi:10.1073/pnas.2410934122
[2] Kong S, Swofford DL, and Kubatko LS. (2024) Inference of phylogenetic networks from sequence data using composite likelihood. Systematic Biology 74:1 53–69 doi:10.1093/sysbio/syae054
[3] Kubatko L, Kong S, Webb E, and Chen Z. (2025) The promise of composite likelihood for species-level phylogenomic inference. Evolutionary Journal of the Linnean Society kzaf008 doi:10.1093/evolinnean/kzaf008